Γενετικές ασθένειες
ορισμός
Μια γενετική ασθένεια ή κληρονομική ασθένεια είναι μια ασθένεια που προκαλείται από ένα ή περισσότερα γονίδια του ενδιαφερόμενου. Το DNA δρα εδώ ως άμεση αιτία της νόσου. Για τις περισσότερες γενετικές ασθένειες, οι αιτιολογικές θέσεις γονιδίων είναι γνωστές. Εάν υπάρχει υποψία γενετικής ασθένειας, η αντίστοιχη διάγνωση μπορεί επομένως να γίνει μέσω γενετικής εξέτασης.
Από την άλλη πλευρά, υπάρχουν επίσης ορισμένες ασθένειες των οποίων η εμφάνιση έχει γενετική επίδραση ή συζητείται, όπως ο σακχαρώδης διαβήτης («διαβήτης»), η οστεοπόρωση ή η κατάθλιψη. Αυτές είναι οι λεγόμενες διαθέσεις, δηλαδή αυξημένη πιθανότητα ορισμένων ασθενειών. Οι διαθέσεις πρέπει να διακρίνονται από κληρονομικές ασθένειες.

Αυτές είναι κοινές κληρονομικές ασθένειες
Σε απόλυτους όρους, οι κληρονομικές ασθένειες δεν είναι συχνές, αλλά οι κληρονομικές ασθένειες που αναφέρονται εδώ εμφανίζονται συχνά σε σύγκριση με άλλες ασθένειες γενετικής αιτίας.
-
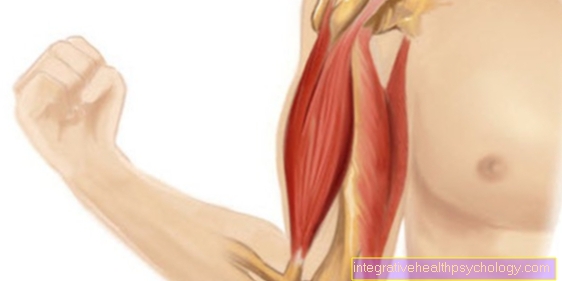
Σύνδρομο Marfan
-
Αναιμία δρεπανοκυττάρων
-
Αιμοφιλία (αιμοφιλία Α ή Β)
-
Παράγοντας V Leiden μετάλλαξη και προκύπτουσα αντίσταση APC
-
Κόκκινο πράσινο αδυναμία
-
Ανεπάρκεια αφυδρογονάσης 6-φωσφορικής γλυκόζης (ανεπάρκεια G6PD)
-
Polydactyly ("πολλαπλά δάχτυλα", επίσης δυνατό ως σύμπτωμα σε άλλες ασθένειες)
-
Τρισωμία 21 (σύνδρομο Down)
-
Chorea Χάντινγκτον
αιτίες
Οι κληρονομικές ασθένειες είναι εξαιρετικά διαφορετικές στην εμφάνισή τους. Βασικά έχουν μόνο ένα κοινό πράγμα: Η αιτία για καθένα από αυτά έγκειται στο DNA, δηλαδή στο γενετικό υλικό του ενδιαφερόμενου. Εδώ μπορούν να προκύψουν διάφορες αλλαγές, όπως μεταλλάξεις (ανταλλαγή πληροφοριών DNA) ή διαγραφές (έλλειψη συγκεκριμένου γενετικού υλικού).
Μεγάλη ποσότητα πληροφοριών κωδικοποιείται στο γενετικό υλικό, όπως τα «σχεδιαγράμματα» για διάφορα συστατικά που είναι σημαντικά για τη λειτουργία ενός κυττάρου του σώματος. Αυτά μπορεί να είναι για παράδειγμα ένζυμα, κανάλια ηλεκτρολυτών ή ουσίες αγγελιοφόρου. Αυτά τα μικρότερα στοιχεία στη συνέχεια διαβάζονται λανθασμένα ή καθόλου από το DNA, το οποίο στη συνέχεια λείπει στο εξελιγμένο σύστημα του σώματος. Η λανθασμένη ή ελλιπής γενετική πληροφορία προκαλεί επομένως ορισμένες δυσλειτουργίες στο σώμα. Αυτά στη συνέχεια προκαλούν συμπτώματα σύμφωνα με το λειτουργικό σύστημα στο οποίο ένα στοιχείο λείπει τώρα.
Μάθετε τα πάντα για το θέμα εδώ: Το γενετικό τεστ.
Έτσι κληρονομούνται οι κληρονομικές ασθένειες
Κάθε κληρονομική ασθένεια κληρονομείται είτε μονογενετικά είτε πολυγενετικά: Αυτό σημαίνει ότι υπάρχουν μία ή περισσότερες γενετικές θέσεις που πρέπει να αλλάξουν για να οδηγήσουν σε μια ασθένεια.
Επιπλέον, τα γενετικά χαρακτηριστικά μπορούν πάντα να κληρονομηθούν με κυρίαρχο ή υπολειπόμενο τρόπο: Το υπολειπόμενο σημαίνει ότι πρέπει να υπάρχει προδιάθεση για τη συγκεκριμένη κληρονομική ασθένεια τόσο στα πατρικά όσο και στα μητρικά γονίδια. Στην περίπτωση της κυρίαρχης κληρονομιάς, μία αλλαγή (δηλαδή ένας γονέας) αρκεί για να προκαλέσει την ασθένεια. Επομένως, με τις κυρίαρχες κληρονομικές ασθένειες, τα άτομα που είναι φορείς θα αρρωστήσουν - ενώ με μια υπολειπόμενη κληρονομιά, συνήθως δεν είναι καν γνωστό ότι υπάρχει αντίστοιχη γενετική προδιάθεση.
Υπάρχουν επίσης ασθένειες που κληρονομούνται μέσω των χρωμοσωμάτων του φύλου, όπως η αιμοφιλία ή η κόκκινη-πράσινη τύφλωση. Οι εγκαταστάσεις για αυτό είναι συνήθως στο χρωμόσωμα Χ, δεδομένου ότι το χρωμόσωμα Υ είναι πολύ μικρό συνολικά και γενικά μπορεί να αποθηκεύσει λίγες γενετικές πληροφορίες. Γι 'αυτό μιλάμε για κληρονομικές ασθένειες που συνδέονται με Χ. Αυτά συνήθως επηρεάζουν σημαντικά περισσότερους άνδρες από τις γυναίκες, καθώς οι γυναίκες μπορούν να αντισταθμίσουν τυχόν εσφαλμένες πληροφορίες σχετικά με το Χ χρωμόσωμα με το δεύτερο.
Το πώς κληρονομείται ακριβώς μια γενετική ασθένεια είναι συνήθως εύκολο να ερευνηθεί αν σας ενδιαφέρει.
Δοκιμές πριν από τη γέννηση
Κατ 'αρχήν, το γενετικό υλικό του παιδιού μπορεί ήδη να εξεταστεί στη μήτρα για όλες τις κληρονομικές ασθένειες των οποίων οι αιτιώδεις γενετικές θέσεις είναι γνωστές. Ωστόσο, οι γενετικές αναλύσεις είναι χρονοβόρες, οπότε συνήθως αναλύεται μόνο η ύποπτη θέση γονιδίου - γι 'αυτό, με τη σειρά του, πρέπει να υπάρχει δικαιολογημένη υποψία γενετικής ασθένειας.
Για μια τέτοια εξέταση, το γενετικό υλικό μπορεί στη συνέχεια να ληφθεί από το αμνιακό υγρό ή τον πλακούντα και να χρησιμοποιηθεί για την ανάλυση.
Ωστόσο, πρέπει πάντα να έχουμε κατά νου ότι οποιαδήποτε επεμβατική διάγνωση συνεπάγεται επίσης κίνδυνο για τη ζωή του αγέννητου παιδιού. Τέτοια τρυπήματα πρέπει επομένως να ζυγίζονται ξεχωριστά σε κάθε περίπτωση.
Υπάρχουν επίσης μετρήσεις που μπορούν να δείξουν μια γενετική ασθένεια, όπως η μέτρηση της νυχικής διαφάνειας ως ένδειξη τρισωμίας 21. Τέτοιες μέθοδοι δεν είναι επικίνδυνες για το αγέννητο παιδί, αλλά δεν μπορούν να προσφέρουν απόλυτη βεβαιότητα σχετικά με την παρουσία μιας γενετικής ασθένειας. Και εδώ, επίσης, μια λειτουργία πρέπει να εξεταστεί προσεκτικά.
Τρισωμία 21
Η αιτία της τρισωμίας 21 είναι το χρωμόσωμα 21, το οποίο δεν υπάρχει δύο φορές αλλά τρεις φορές στα προσβεβλημένα άτομα. Αυτή η παραλλαγή του DNA δημιουργείται όταν τα χρωμοσώματα κατανέμονται στα γονικά βλαστικά κύτταρα, δηλ. Στο σπέρμα ή στα ωάρια. Επομένως, είναι "σφάλμα διανομής" και όχι αλλαγή στο πραγματικό γενετικό υλικό. Αυτό εξηγεί γιατί η τρισωμία 21 μπορεί να εμφανιστεί αυθόρμητα σε κάθε οικογένεια και γιατί η πιθανότητα να αποκτήσει παιδί με σύνδρομο Down είναι η ίδια σε όλες τις οικογένειες. Ακριβώς μιλώντας, η τρισωμία 21 - όπως και άλλες τρισωμίες - δεν πρέπει να θεωρηθεί ως κληρονομική ασθένεια με την πραγματική έννοια. Ωστόσο, η τρισωμία 21 είναι η πιο κοινή ασθένεια που σχετίζεται με το DNA στα νεογνά.
Τα χαρακτηριστικά του αλλαγμένου συνόλου χρωμοσωμάτων στο σύνδρομο Down μπορούν ήδη να παρατηρηθούν στο αγέννητο παιδί στη μήτρα: Οι καθυστερήσεις στην ανάπτυξη και τα ελαττώματα μπορούν να οδηγήσουν, μεταξύ άλλων, σε ένα κρανίο που είναι πολύ μικρό, κοντά οστά του μηρού και του άνω βραχίονα και καρδιακά ελαττώματα. Μια μεγάλη ποσότητα αμνιακού υγρού μπορεί επίσης να αποτελεί ένδειξη τρισωμίας 21, καθώς τα προσβεβλημένα αγέννητα παιδιά πίνουν ή καταπίνουν σχετικά λίγο αμνιακό υγρό. Ωστόσο, κανένα από αυτά τα χαρακτηριστικά δεν είναι καθοριστικά σημάδια του συνδρόμου Down!
Εκτός από τα σημάδια επιβράδυνσης της ανάπτυξης που αναφέρθηκαν, τα παιδιά με σύνδρομο Down συχνά εμφανίζουν επίσης καθυστερημένη ανάπτυξη, για παράδειγμα στους τομείς των γλωσσικών και κινητικών δεξιοτήτων. Τα άτομα που πάσχουν από σύνδρομο Down συχνά παρουσιάζουν αξιοσημείωτες κοινωνικές δεξιότητες, ενώ η νοημοσύνη παραμένει συχνά κάτω από τον μέσο όρο. Ωστόσο, τα άτομα που έχουν πληγεί διαφέρουν πολύ σε αυτά τα χαρακτηριστικά και δεν είναι ασυνήθιστο να αποφοιτούν από το σχολείο αφού λάβουν καλή υποστήριξη.
Αργότερα στη ζωή, τα άτομα με τρισωμία 21 έχουν αυξημένο κίνδυνο διάγνωσης με ορισμένες ασθένειες. Αυτές περιλαμβάνουν τη νόσο του Αλτσχάιμερ, την επιληψία και τον καρκίνο, ιδιαίτερα τη λευχαιμία. Ωστόσο, το προσδόκιμο ζωής των ατόμων με σύνδρομο Down συνεχίζει να αυξάνεται: Εν τω μεταξύ, τα προσβεβλημένα άτομα φτάνουν συχνά στην ηλικία των 60 ή 70 ετών.
Μπορείτε να βρείτε περισσότερες πληροφορίες στον ιστότοπό μας Κάτω σύνδρομο
Ανεπάρκεια άλφα-1 αντιτρυψίνης
Η ανεπάρκεια άλφα-1 αντιτρυψίνης μπορεί να έχει διάφορες μορφές και μορφές, ανάλογα με τα ακριβή γενετικά χαρακτηριστικά του ατόμου που πάσχει. Αυτό σημαίνει ότι δεν είναι κάθε ανεπάρκεια της αντιπρυψίνης άλφα-1 σε συμπτώματα. Στη συνέχεια, θα συζητηθεί μόνο ο κλινικά εμφανής τύπος (PiZZ) αυτής της γενετικά προσδιορισμένης νόσου.
Το ενζυμικό ελάττωμα που υπάρχει σε αυτήν την ασθένεια προκαλεί τη διάσπαση και την αναδιαμόρφωση των δομικών μονάδων στον ιστό των οργάνων σε προσβεβλημένα άτομα. Επιπλέον, οι ελαττωματικές πρωτεΐνες διηθούνται από το αίμα από το ήπαρ και συσσωρεύονται εκεί. Αυτό μπορεί να οδηγήσει σε φλεγμονή του ήπατος (ηπατίτιδα), κίρρωση ή καρκίνο του ήπατος. Οι αεραγωγοί στους πνεύμονες γίνονται ασταθείς λόγω της έλλειψης σταθερού ιστού και καταρρέουν ταχύτερα: Αναπτύσσεται η κλινική εικόνα της ΧΑΠ (χρόνια αποφρακτική πνευμονοπάθεια). Αυτή η κλινική εικόνα είναι συχνά το πρώτο σύμπτωμα της ανεπάρκειας της αντιπρυψίνης άλφα-1, οπότε κάθε άτομο με ΧΑΠ σε μικρότερη ηλικία θα πρέπει να ελέγχεται για ανεπάρκεια της άλφα-1 αντιτρυψίνης.
Εάν η ασθένεια έχει παραμείνει για μεγάλο χρονικό διάστημα, οι πνεύμονες μπορεί να υπερχειλίσουν, καθώς ο αέρας που αναπνέετε δεν μπορεί να εκπνεύσει σωστά μέσω των ασταθών αεραγωγών και να συσσωρευτεί στους πνεύμονες. Ως θεραπεία, εκτός από τη συνεχή αποφυγή του καπνίσματος τσιγάρων και τακτικούς εμβολιασμούς για την πρόληψη αναπνευστικών παθήσεων, θα πρέπει επίσης να ληφθούν φαρμακευτικά μέτρα: Η ελλείπουσα άλφα-1-αντιτρυψίνη μπορεί να χορηγηθεί ενδοφλεβίως προκειμένου να ανακουφίσει τα συμπτώματα όσο το δυνατόν περισσότερο και να σταματήσει την πορεία της νόσου.
Μπορείτε να βρείτε περισσότερες πληροφορίες στον ιστότοπό μας Ανεπάρκεια άλφα-1 αντιτρυψίνης
αιμοφιλία
Η ομάδα της αιμοφιλίας είναι επίσης γνωστή ως «αιμοφιλία», καθώς αυτός ο όρος περιγράφει με ακρίβεια το κύριο σύμπτωμα αυτής της κληρονομικής νόσου: οι πάσχοντες αιμορραγούν περισσότερο και, ανάλογα με τη σοβαρότητα της νόσου, πιο συχνά από ό, τι δεν επηρεάζονται.
Η αιμορραγία σταματά συνήθως από αυτό που είναι γνωστό ως καταρράκτη πήξης, ένα ενδογενές μονοπάτι σηματοδότησης που αποτρέπει την υπερβολική απώλεια αίματος. Σε αυτό το σύστημα πήξης, 13 παράγοντες παίζουν ρόλο, οι οποίοι ενεργοποιούν ο ένας τον άλλον μετά τον άλλο. Αυτό μπορεί να φανταστεί ως μια σειρά ντόμινο: εάν χτυπήσετε μια πέτρα (παράγοντας πήξης), ενεργοποιείται η επόμενη και ούτω καθεξής. Στο τέλος αυτής της διαδρομής σήματος ή των ντόμινο υπάρχει πήξη του αίματος. Με την αιμορροφιλία, λείπει ένας συγκεκριμένος παράγοντας - ανάλογα με τον συγκεκριμένο υποτύπο της νόσου: η αλυσιδωτή αντίδραση διακόπτεται εδώ.
Η θεραπεία για την ασθένεια μπορεί να πραγματοποιηθεί προσδιορίζοντας τον παράγοντα που λείπει και προσθέτοντάς τον από έξω. Τα προσβεβλημένα άτομα πρέπει επομένως να εγχέονται τακτικά με ένα παρασκεύασμα με αυτόν τον παράγοντα πήξης έτσι ώστε να μπορεί να λάβει χώρα η υπόλοιπη αλυσιδωτή αντίδραση.
Μπορείτε να βρείτε περισσότερες πληροφορίες στον ιστότοπό μας Ασθένεια αίματος
Κυστική ίνωση
Στη γενετική νόσο η κυστική ίνωση - επίσης γνωστή ως κυστική ίνωση - υπάρχει ελαττωματική παραγωγή καναλιών ιόντων, πιο συγκεκριμένα των καναλιών χλωρίου. Ως αποτέλεσμα, η σύνθεση των εκκρίσεων του σώματος (π.χ. ιδρώτας, εκκρίσεις από την αναπνευστική οδό και το πάγκρεας) των προσβεβλημένων ατόμων αλλάζει: Δεδομένου ότι η έλλειψη χλωριδίου σημαίνει ότι εισέρχεται λιγότερο νερό στον αγωγό του αντίστοιχου αδένα, η έκκριση είναι σχετικά ιξώδης.
Ως αποτέλεσμα, τα συμπτώματα συνήθως αναπτύσσονται στον πεπτικό σωλήνα, καθώς η έκκριση με τα πεπτικά ένζυμα δεν μπορεί να ρέει καλά από το πάγκρεας στο έντερο και έτσι βλάπτει το ίδιο το πάγκρεας. Επιπλέον, είναι συχνές οι πεπτικές διαταραχές όπως τα λιπαρά κόπρανα, η διάρροια και το προκύπτον χαμηλό σωματικό βάρος.
Η δεύτερη μεγάλη ομάδα συμπτωμάτων αναπτύσσεται συνήθως στους πνεύμονες: Δεδομένου ότι η βλέννα που εμφανίζεται φυσικά στους πνεύμονες είναι πιο ιξώδης από ό, τι σε υγιείς ανθρώπους, είναι πιο δύσκολο να την αφαιρέσετε από τους βλεφαρίδες. Αυτό μπορεί να οδηγήσει σε χρόνιο βήχα και απόφραξη των βρόγχων (βρογχιεκτασία). Η μεγαλύτερη ποσότητα έκκρισης των πνευμόνων παρέχει επίσης ένα καλό περιβάλλον για την ανάπτυξη βακτηρίων, με αποτέλεσμα συχνές αναπνευστικές λοιμώξεις και πνευμονία.
Η κυστική ίνωση αντιμετωπίζεται συμπτωματικά με αποχρεμπτικά, πεπτικά ένζυμα και αντιβιοτικά για λοιμώξεις.
Μπορείτε να βρείτε περισσότερα σχετικά με αυτό στον ιστότοπό μας Κυστική ίνωση
Factor V Leiden και APC Αντίσταση
Η μετάλλαξη του παράγοντα V Leiden περιλαμβάνει μια αλλαγή στις γενετικές πληροφορίες που μπορεί να προκαλέσει αυξημένη πήξη του αίματος. Ο λόγος για αυτό είναι ο παράγοντας V στον λεγόμενο καταρράκτη πήξης του σώματος: αυτή η διαδρομή σήματος διασφαλίζει ότι σε περίπτωση τραυματισμού, το τραύμα κλείνει από τις "κολλητικές πρωτεΐνες" του ίδιου του σώματος (ινώδες). Υπάρχουν 13 παράγοντες σε αυτό το μονοπάτι σηματοδότησης, οι οποίοι ονομάζονται με λατινικούς αριθμούς (σημαίνει "Παράγοντας 5 που υποφέρει"!). Ο παράγοντας V έχει ευεργετική επίδραση στο σχηματισμό βύσματος ινώδους, αλλά μπορεί επίσης να ανασταλεί από τη λεγόμενη ενεργοποιημένη πρωτεΐνη C (APC για συντομία). Αυτό παίζει σημαντικό ρόλο στη ρύθμιση αυτής της οδού σηματοδότησης και στην πρόληψη της υπερβολικής πήξης του αίματος.
Ο μεταλλαγμένος παράγοντας V υπάρχει στα προσβεβλημένα άτομα αλλά δεν αποκρίνεται στο APC. Το σώμα δεν διαθέτει μια σημαντική «συσκευή ασφαλείας» σε αυτό το σημείο για να αποτρέψει την πήξη του αίματος χωρίς λόγο, η οποία μπορεί ακόμη και να εμποδίσει τα αγγεία και ως εκ τούτου να προκαλέσει κυκλοφορικές διαταραχές.
Στατιστικά, τα άτομα που επηρεάζονται από τη μετάλλαξη του παράγοντα V Leiden είναι πιο πιθανό να παρουσιάσουν θρομβωτικό συμβάν (δηλαδή θρόμβωση ή πνευμονική εμβολή), ακόμη και χωρίς ιστορικό τυπικών παραγόντων κινδύνου. Σε τεχνικούς όρους, κάποιος μιλά επίσης για «θρομβοφιλία», δηλαδή την τάση για πήξη.
Μπορείτε να βρείτε περισσότερα σχετικά με αυτό στον ιστότοπό μας Παράγοντας V Leiden
Νόσος του Gaucher
Στη νόσο του Gaucher, η αλλαγή στις πληροφορίες του DNA προκαλεί ελάττωμα σε ένα ένζυμο που εμπλέκεται στον μεταβολισμό των λιπιδίων, πιο συγκεκριμένα στη γλυκοκερεοβροσιδάση: Αυτό βοηθά στη διάσπαση των παλαιών κυτταρικών συστατικών. Σε περίπτωση ελαττώματος, μπορεί να υπάρξει μείωση της λειτουργικότητας ή ακόμη και απώλεια της λειτουργικότητας, και κατά συνέπεια τα συμπτώματα εμφανίζονται στην παιδική ηλικία ή την ενηλικίωση.
Τα συμπτώματα της νόσου Gaucher οφείλονται σε μεγάλο βαθμό στη διεύρυνση του ήπατος και του σπλήνα, η ανάπτυξη της οποίας ο οργανισμός προσπαθεί να αντισταθμίσει την έλλειψη ενζύμων. Αυτό αυξάνει την ανάλυση όλων των συστατικών του αίματος, τα οποία μπορούν να αναγνωριστούν στον αριθμό αίματος και να χρησιμοποιηθούν ως διαγνωστικός δείκτης μαζί με το διευρυμένο ήπαρ και σπλήνα.
Το ένζυμο που λείπει γλυκοκερεοβροσιδάση μπορεί να χρησιμοποιηθεί θεραπευτικά ως φάρμακο. Η πρόγνωση και η πορεία της νόσου του Gaucher εξαρτάται σε μεγάλο βαθμό από τη σοβαρότητα της απώλειας λειτουργίας του ενζύμου.
Για περισσότερες πληροφορίες, διαβάστε εδώ: Η νόσος του Gaucher.
Η νόσος του Όσλερ
Η νόσος του Osler είναι μια κληρονομική ασθένεια που χαρακτηρίζεται από έντονη αγγειοδιαστολή. Κατ 'αρχήν, αυτή η επέκταση των αγγείων μπορεί να συμβεί οπουδήποτε, τόσο στο δέρμα όσο και στα εσωτερικά όργανα. Τα τοιχώματα των διευρυμένων αγγείων είναι σχετικά λεπτά και εύκολα σκίζονται. Ως αποτέλεσμα, οι πληγείσες περιοχές αιμορραγούν γρήγορα.
Η αγγειοδιαστολή εμφανίζεται ιδιαίτερα συχνά στο πρόσωπο και στη ρινική βλεννογόνο μεμβράνη, οπότε οι πάσχοντες συνήθως παραπονούνται για συχνές ρινορραγίες και μικρή κηλίδα αιμορραγίας στο πρόσωπο.
Εάν υπάρχει υποψία για τη νόσο του Osler, θα πρέπει να πραγματοποιούνται κατάλληλα διαγνωστικά, καθώς η αγγειοδιαστολή μπορεί επίσης να συμβεί σε ζωτικά όργανα ή όργανα με καλή παροχή αίματος, όπως οι πνεύμονες, ο εγκέφαλος ή το ήπαρ, στα οποία η αιμορραγία από ένα ρήξη αγγείου είναι επικίνδυνη.
Μπορείτε να βρείτε περισσότερα σχετικά με αυτό το θέμα στον ιστότοπό μας Η νόσος του Όσλερ
Η νόσος του Recklinghausen
Η νευροϊνωμάτωση τύπου 1 - ή η νόσος του Recklinghausen - είναι μια γενετική ασθένεια στην οποία όσοι επηρεάζονται συχνά αναπτύσσουν όγκους στα κύτταρα του νευρικού καλύμματος. Οι όγκοι που αναπτύσσονται μπορεί να είναι καλοήθεις και κακοήθεις και εμφανίζονται σε νεαρή ηλικία.
Οι τυπικοί όγκοι, ωστόσο, είναι καλοήθεις νευροϊστοί: Αυτοί αποτελούνται από κύτταρα που περιβάλλουν και απομονώνουν το νεύρο σαν ηλεκτρικό καλώδιο, καθώς και από τον περιβάλλοντα συνδετικό ιστό. Είναι καλοήθεις, δηλαδή μη-εξαπλωμένοι και αργά αναπτυσσόμενοι όγκοι.
Ωστόσο, η χειρουργική επέμβαση για την απομάκρυνση των νευροϊνών μπορεί να είναι δύσκολη, καθώς συχνά συνδέονται σταθερά στο νεύρο και στη συνέχεια πρέπει να αφαιρεθεί το αντίστοιχο νεύρο. Παρ 'όλα αυτά, αυτή είναι η μόνη θεραπευτική επιλογή για συμπτωματικό νευροϊύρωμα, καθώς η αιτιώδης θεραπεία για αυτήν την κληρονομική νόσο δεν είναι δυνατή.
Μπορείτε να βρείτε περισσότερα σχετικά με αυτό το θέμα στον ιστότοπό μας Νευροϊνωμάτωση τύπου 1
Μυική δυστροφία
Ο όρος μυϊκή δυστροφία περιγράφει μια ομάδα κληρονομικών ασθενειών στις οποίες ορισμένα συστατικά των μυών δεν μπορούν ή δεν μπορούν να συναρμολογηθούν σωστά από τα κύτταρα του σώματος. Ως αποτέλεσμα, οι πάσχοντες αναπτύσσουν συνήθως μυϊκή αδυναμία ήδη από την παιδική ηλικία και την εφηβεία, και αυτό μπορεί να οδηγήσει σε απώλεια μυϊκής μάζας, περιορισμούς κίνησης και ακόμη και σωματικές αναπηρίες.
Εάν υπάρχει υποψία παρουσίας μυϊκής δυστροφίας, πρέπει πρώτα να προσδιοριστούν οι τιμές του αίματος. Εάν οι τιμές ταιριάζουν με την υποψία διάγνωσης, μπορεί ακόμη να πραγματοποιηθεί μυϊκή βιοψία: Λαμβάνεται ένα μικρό δείγμα ιστού από τον μυ, ο οποίος στη συνέχεια εξετάζεται μικροσκοπικά για κυτταρικά ελαττώματα. Μια γενετική εξέταση είναι επίσης δυνατή για τη διαπίστωση της διάγνωσης, καθώς οι αντίστοιχες γενετικές θέσεις είναι συνήθως γνωστές για τις διάφορες μορφές μυϊκής δυστροφίας και θα έπρεπε να αλλάξουν. Η αιτιώδης θεραπεία για μυϊκές δυστροφίες δεν είναι γνωστή.
Μπορείτε να βρείτε περισσότερα σχετικά με αυτό το θέμα στον ιστότοπό μας Μυική δυστροφία
Xeroderma pigmentosum
Το Xeroderma pigmentosum είναι μια σπάνια κληρονομική ασθένεια στην οποία ορισμένα ένζυμα στο δέρμα του προσβεβλημένου ατόμου δεν λειτουργούν. Αυτά τα ένζυμα κανονικά φροντίζουν την επιδιόρθωση του DNA, το οποίο μπορεί να υποστεί βλάβη από το φως του ήλιου ή το περιέχον φως UVB. Η βλάβη UVB μπορεί να προκαλέσει καρκίνο του δέρματος σε άτομα που έχουν προσβληθεί καθώς και σε όλους τους άλλους ανθρώπους, αλλά με το Xeroderma Pigmentosum η διαδικασία επιταχύνεται λόγω της έλλειψης μηχανισμών επισκευής. Ως αποτέλεσμα, οι πάσχοντες αναπτύσσουν σοβαρές μορφές καρκίνου του δέρματος στην παιδική ηλικία και την εφηβεία και μετά από μια σύντομη έκθεση στο ηλιακό φως.
Η αιτιώδης θεραπεία δεν είναι ακόμη δυνατή. Οι πληγέντες πρέπει να αποφύγουν το φως του ήλιου για όλη τη ζωή, γι 'αυτό το ψευδώνυμο «παιδιά σεληνόφωτου» έχει καθιερωθεί για τους πληγέντες (μερικές φορές πολύ νέους). Επιπλέον, αυτά τα άτομα πρέπει να παρακολουθούνται από έναν δερματολόγο για τακτική εξέταση καρκίνου του δέρματος, προκειμένου να απομακρυνθεί αμέσως ο καρκίνος του δέρματος που αναπτύχθηκε πρόσφατα. Εάν ακολουθούνται αυστηρά αυτά τα μέτρα, το προσδόκιμο ζωής ενός ατόμου με xeroderma pigmentosum είναι περίπου το ίδιο με αυτό ενός μη επηρεασμένου ατόμου.
Μπορείτε να βρείτε περισσότερα για αυτήν την ασθένεια στον ιστότοπό μας Xeroderma pigmentosum
Σύνδρομο Lynch
Το σύνδρομο Lynch είναι μια αλλαγή στο DNA που προκαλεί ελαττωματικό ένζυμο στα κύτταρα του σώματος.Στα προσβεβλημένα άτομα, ένας συγκεκριμένος μηχανισμός είναι επομένως ελαττωματικός, ο οποίος διαφορετικά υποτίθεται ότι προστατεύει τα κύτταρα από εκφυλισμό, δηλαδή ανεξέλεγκτη ανάπτυξη - τα άτομα με σύνδρομο Lynch έχουν συνεπώς πολύ αυξημένο κίνδυνο εμφάνισης καρκίνου.
Ο καρκίνος του παχέος εντέρου συμβαίνει συχνά επειδή τα κύτταρα φυσικά χωρίζονται εδώ ούτως ή άλλως και τα σφάλματα στον προγραμματισμό ανάπτυξης και θανάτου ενός κυττάρου γίνονται πιο γρήγορα εμφανή. Τα προσβεβλημένα άτομα αναπτύσσουν συχνά έναν όγκο στο παχύ έντερο σε ασυνήθιστα νεαρή ηλικία, δηλαδή πριν από την ηλικία των 50 ετών, το οποίο στη συνέχεια ονομάζεται HNPCC (κληρονομικός μη πολύποδος καρκίνος του παχέος εντέρου). Ωστόσο, δεν θα αναπτύξουν καρκίνο του παχέος εντέρου όλοι όσοι έχουν τη γενετική σύνθεση του συνδρόμου Lynch. Από την άλλη πλευρά, άλλα όργανα μπορούν επίσης να αναπτύξουν έναν όγκο, καθώς οι γενετικές προθέσεις που ευνοούν την ανάπτυξη ενός όγκου υπάρχουν σε όλα τα κύτταρα του σώματος. Επομένως, είναι απαραίτητοι τακτικοί έλεγχοι και προληπτικές εξετάσεις για όσους πάσχουν από σύνδρομο Lynch, προκειμένου να αντιμετωπιστούν επαρκώς οι όγκοι που αναπτύσσονται σε πρώιμο στάδιο.
Μπορείτε να βρείτε περισσότερα σχετικά με αυτό το θέμα στον ιστότοπό μας Σύνδρομο Lynch